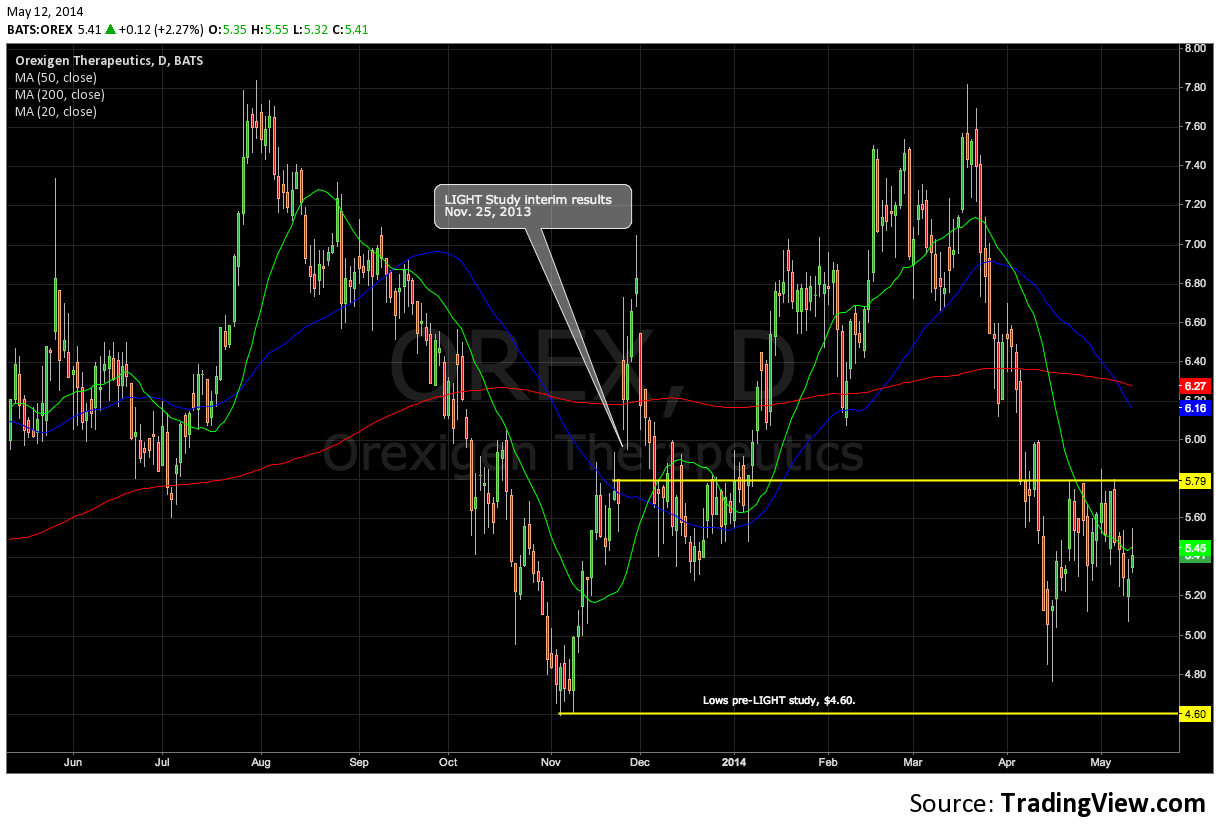
- Stock has retraced 30% from highs early this year as air is let out of many lofty valuations in biotech, and now trades at prices that pre-date interim results from cardiovascular outcomes trial, the LIGHT study.
- High likelihood that Orexigen has assuaged early FDA concerns and NB32 is approved in early June.
Orexigen Therapeutics (OREX) resubmitted the New Drug Application for lead asset and weight-loss drug, NB32 (naltrexone/bupropion sustained release), in December of 2013, setting up a value-driving event for June 10th with the FDA’s PDUFA goal date. Orexigen is 30% off of recent highs on broad sector weakness, and we like the fact that the equity has gotten significantly cheaper in front of a key inflection point. We’ve watched OREX fall since late March and believe investors can be comfortable owning the stock at prices below where the stock traded prior to the LIGHT study results.
Although obesity treatments from competitors Vivus (VVUS) and Arena Pharmaceuticals (ARNA) have not performed to expectations from Wall Street (revenues of $23.7M TTM and $30M run rate, respectively), investors seem willing to give Orexigen the benefit of the doubt due to NB32’s clinical profile, the presence of a formidable marketing partner in Takeda Pharmaceuticals, and results from a large, controlled study of the drug’s effects on cardiovascular safety. Many point to Orexigen as the “third time’s a charm” player in the prescription weight loss environment, which is led by Arena at a valuation of $1.5 billion, and Vivus at $540 million. We believe NB32 will be approved next month, making it an attractive equity to own in the short-term.
In this environment, we’re not looking for a homerun; owning OREX into the June 10th PDUFA decision and booking gains on the approval makes sense. Those expecting OREX to trade at parity with ARNA following the FDA’s decision are likely to be disappointed, and selling the approval will be a popular trade. We note that accounting for convertibles and options, OREX’s fully diluted share count is nearly 45% higher than its outstanding. Investors must consider this $875M fully diluted market cap, not the $600M apparent at a glance.
Cardiovascular Data Increases Likelihood of Approval
Given historical comparisons to Vivus and Arena, in addition to the SPA for the LIGHT trial, we expect NB32 to receive FDA approval in June.
Orexigen is treading a strikingly similar path to market as approved weight-loss products marketed by Vivus (Qsymia) and Arena Pharmaceuticals (Belviq): 1) file original NDA; 2) FDA advisory panel; 3) CRL noting requirement for more long-term data; 4) address FDA concerns; 5) re-submit NDA; 6) another advisory panel held; 7) subsequent approval.
There are a few distinct differences in Orexigen’s case that bode well for investors, namely the company is proceeding with its phase 3 trial under an SPA and a second advisory panel is unlikely this late in the game.
Orexigen’s CRL in early 2011 noted a single deficiency related to proof of cardiovascular safety in a population of obese patients. Orexigen met with the FDA and agreed to a large phase 3 trial to rule out an increase in the risk of major adverse cardiovascular events (MACE) in patients on NB32 compared to placebo. The LIGHT Study (protocol) is a 10,000-patient randomized, placebo-controlled study comparing NB32 to placebo on MACE rate such as heart attack or stroke in overweight and obese patients. The FDA and Orexigen agreed to the trial design under a Special Protocol Assessment. The FDA concluded in its SPA with Orexigen that a positive interim analysis from the LIGHT Study would be sufficient, in combination with data from previous registration trials, for approval of NB32.
Orexigen announced in November of 2013 that an interim analysis of the LIGHT Study achieved the FDA’s pre-specified criteria – exclude a hazard ratio of 2.0, using the upper bound of the 95% confidence interval, for excess risk of major adverse cardiovascular events – and the company resubmitted its NDA in December of 2013. The independent Data Monitoring Committee’s summary report of the interim analysis from the study formed the basis of the NDA resubmission, which was supplied to the FDA in February of 2014.
While Orexigen has not released detailed results from the LIGHT study, management has been up-front in investor communications trumpeting very positive results and expectations of FDA and EU approval. We are encouraged that Orexigen will be the only weight-loss drug company providing a cardiovascular outcomes trial to the FDA prior to approval. Neither Vivus nor Arena undertook specific trials prior to approval in the U.S., or application in the EU.
Our one point of contention is NB32’s higher relative rate of patient discontinuation in previous trials due to adverse events. A pooled 23.8% of patients across registration trials discontinued due to adverse events. We note the higher rate of discontinuation, which is prevalent in many obesity drug trials, but are comforted that the FDA had this information in hand with the first NDA submission and did not express concern in its CRL. We view discontinuation rates as a sticking point, but ultimately, one that likely won’t influence an FDA decision this time around. Development under an SPA and achievement of the goals at interim significantly de-risk the possibility of a second CRL.
Recall that both Vivus’ Qsymia and Arena’s Belviq faced a second advisory panel following re-submission of their respective NDA’s, a risk mitigated for Orexigen by the proximity of this PDUFA decision and no indication of an advisory panel to date. Panel meetings are typically announced in the 3-4 months following NDA submission. Orexigen’s NDA was resubmitted on December 11th, thus the sweet spot passed in early April.
Considering Events Post-Approval
Beyond June, news-flow centered on an EMA decision, milestone payments from Takeda, and a possible ex-U.S. partnership will take the spotlight.
On February 25, Orexigen announced that in response to the company’s European marketing application for NB32, submitted in October of 2013, it had received the Day 120 List of Questions from the EU’s Committee for Medicinal Products for Human Use (CHMP). According to the company, the EMA reviewers’ questions were consistent with the issues raised by the FDA during the course of NB32 development and the initial review of the NDA, “such as manufacturing and quality, non-clinical studies, dosing regimen, and, most significantly, cardiovascular safety.” Orexigen expects to address the issues and reply by the end of May. Most importantly, Orexigen is providing EMA reviewers with the interim results of the LIGHT trial to assuage cardiovascular safety concerns, a stringent hurdle at the EMA.
Orexigen has been oddly effusive with investors about expectations in Europe, with CEO Michael Narachi “highly confident” in a positive opinion from the CHMP. Making these comments more interesting is the fact that European regulators have not yet seen the interim results of the LIGHT study: this summary will be submitted with Orexigen’s response to the Day 120 List of Questions. Is Narachi being overly optimistic, or have regulators in Europe indicated that the interim LIGHT results, similar to the FDA, are sufficient for approval?
This decision is likely in the third quarter, with news flow on later stages of the EMA process (list of outstanding issues/oral explanation) commanding investor attention in 2H14.
We are optimistic that NB32 will succeed where Qsymia and Belviq fell short across the pond. Orexigen, similar to the U.S., has the benefit of seeing the trial-and-error experiences of Vivus and Arena in Europe. Qsymia was rejected on cardiovascular concerns (lacked CVOT study before filing) while Arena pulled Belviq’s application at the Day 120 Question stage, subsequently selling European rights to Eisai Co., who plans to re-file in 2015. NB32 strikes a more appropriate balance of efficacy and safety compared to Qsymia (efficacious but a lacking safety profile) and Belviq (safer but lacking relative efficacy), and most importantly, has interim results from the LIGHT study in-hand ahead of this decision.
Orexigen had a cash balance of $155M at March 31, 2014, and more importantly, a plethora of post-approval milestone payments from partner Takeda totaling $100M: $20M on approval, $10M on delivery of first commercial goods, and $70M on first sale of the product. All-in, Orexigen should receive $0.90/share ($0.62 on a fully diluted basis), in cash over the 6 months following US approval. Orexigen has guided for operating expenses of $75-85M in 2014. Further, the company receives a tiered royalty on Takeda’s NB32 sales ranging from 20-35%, up to $880M in sales milestones, and anniversary payouts amounting to $45M.
Conclusion
Orexigen has become progressively cheaper over the last two months, and owning the stock in anticipation of its June 10th PDUFA presents an attractive trade in a choppy environment. We would not, however, be surprised to see Orexigen sell-the-news on approval as biotech remains skittish. Investors are better off taking the short-term view with Orexigen. We believe Wall Street has over-estimated the commercial opportunity for prescription weight-loss drugs, and although NB32 is likely to outperform its competitors, we suspect none of these assets will live up to the blockbuster status promoted by analysts as this class of drugs approached commercialization in 2012 and 2013. For this reason, we think there are more attractive long-term opportunities to be had.
Following what we suspect will be a post-approval fade, it’s worth revisiting Orexigen in the 1-3 months after NB32’s approval, ahead of the EMA decision and a potential ex-U.S. partnership.